

JAK2
JAK2 associates with the cytoplasmic regions of numerous cytokine receptors including those for growth hormone, erythropoietin, leptin, interferon gamma, interleukin-3, and interleukin-5. JAKs contain tandem protein kinase domains: a pseudokinase domain (JH2) and a tyrosine kinase domain (JH1).
In collaboration with Olli Silvennoinen's lab in Finland, researchers in the Hubbard Lab showed that the pseuodokinase domain of JAK2 is actually an active protein kinase, despite substitution of several residues that are conserved in canonical protein kinases. We determined a crystal structure of JAK2 JH2, which revealed that this domain adopts the eukaryotic protein kinase fold but binds Mg-ATP more tightly than a canonical protein kinase (see figure 1).
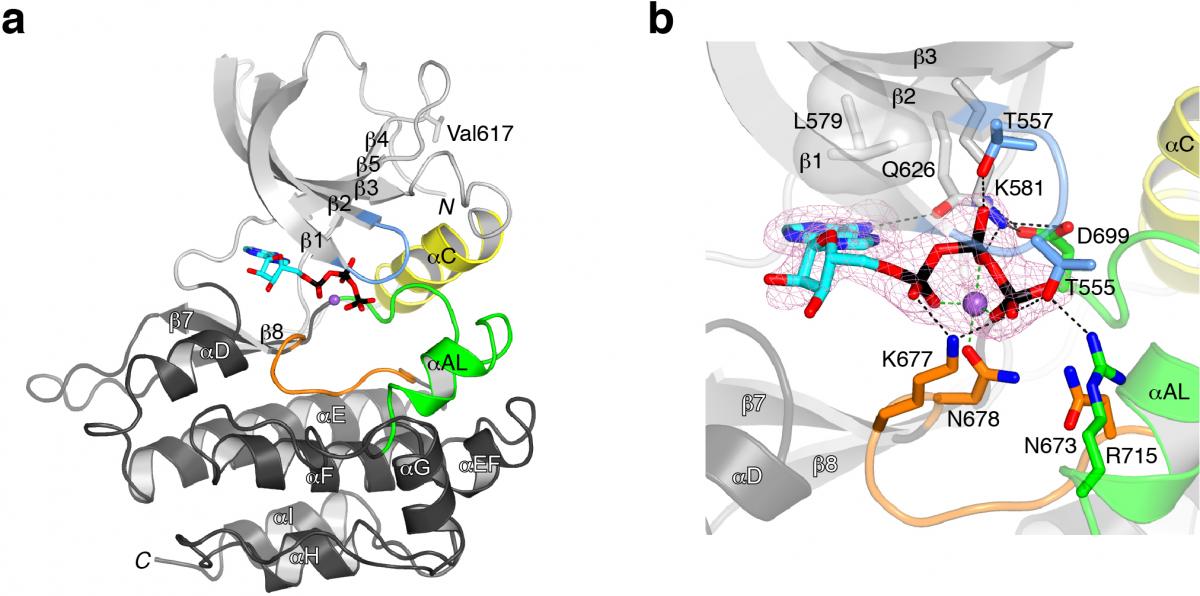
Read more in our paper “Crystal structures of the JAK2 pseudokinase domain and the pathogenic mutant V617F” published in Nature Structural & Molecular Biology.
Numerous mutations in the pseudokinase region of JAK2 result in constitutive tyrosine kinase activity and are causally linked to myeloproliferative neoplasms (MPNs) and leukemias in humans. These and other data indicate that the pseudokinase domain serves an autoinhibitory role in JAK2 regulation. Crystal structures of the individual pseudokinase (see figure 1) and kinase domains of JAK2 have been determined previously, but the structure and organization of the tandem domains is unknown, and therefore the molecular bases for pseudokinase-mediated autoinhibition and pathogenic activation remain obscure.
In collaboration with Yibing Shan at D.E. Shaw Research, we used long time-scale molecular dynamics simulations to generate a structural model for the autoinhibitory interaction between the pseudokinase and kinase domains of JAK2. A striking feature of our model, which is supported by charge-reversal mutagenesis experiments, is that nearly all of the activating disease mutations are present in the pseudokinase–kinase interface (see figure 2). The simulations indicate that the kinase domain is stabilized in an inactive state by the pseudokinase domain, and they offer a molecular rationale for the hyperactivation of the predominant JAK2 MPN mutant, V617F.
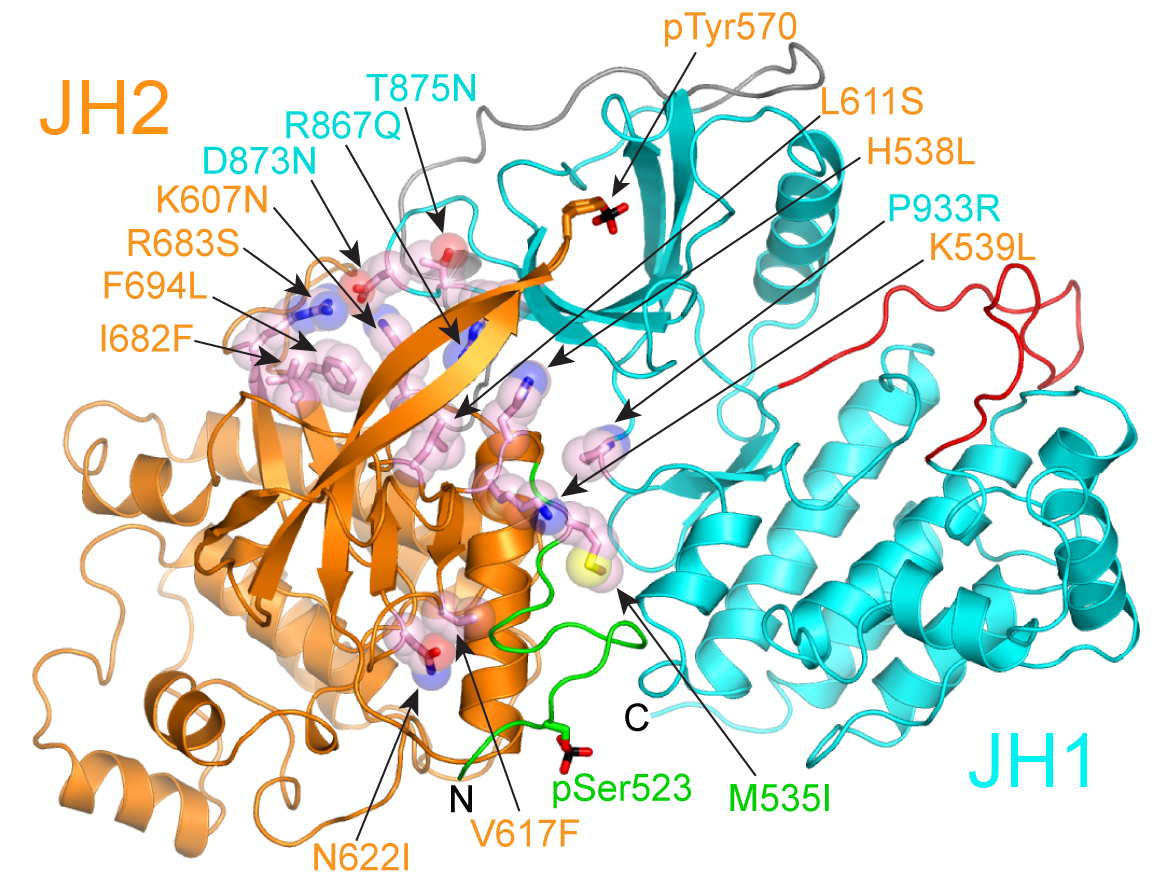
Read more in our paper “Molecular basis for pseudokinase-dependent autoinhibition of JAK2 tyrosine kinase” published in Nature Structural & Molecular Biology.